def normalize_trait_name(name): rename_map = {"Saxena": "Daytime_sleepiness", }ifnotisinstance(name, str):return name name = name.replace("-", "_") name = rename_map.get(name, name)returnstr(name)def get_matrix_df_from_json(fpath, check_sanity=True):withopen(Path(fpath), "r") as f: d = json.load(f) rows = [] traits = []for a_vs_b, val in d.items(): trait_a, trait_b = ([normalize_trait_name(x) for x in a_vs_b.split("_vs_")]) traits.append(trait_a) traits.append(trait_b) rows.append({"trait_a": trait_a,"trait_b": trait_b,"rg": val, }) traits =sorted(list(set(traits))) df = pd.DataFrame(rows) n =len(traits) R = pd.DataFrame(np.nan, index=traits, columns=traits, dtype=float) R.values[R.index.get_indexer(df["trait_a"]), R.columns.get_indexer(df["trait_b"])] = df["rg"].to_numpy() R.values[R.index.get_indexer(df["trait_b"]), R.columns.get_indexer(df["trait_a"])] = df["rg"].to_numpy()if check_sanity:# Max and min valuesprint(f"max={R.max().max():.3g}, min={R.min().min():.3g}")# Symmetry sanity (in case the input accidentally contains BOTH directions) asym = np.nanmax(np.abs(R.values - R.values.T))print(f"max |R - R.T| = {asym:.3g}") # expect 0.0 if each pair listed once# Coverage: how many off-diagonal pairs are actually populated? off =~np.eye(len(traits), dtype=bool)print(f"populated off-diagonal fraction: {np.isfinite(R.values[off]).mean():.3f}")return RR_df = get_matrix_df_from_json(Path(datadir) /"ldsc_results/rg.txt")np.fill_diagonal(R_df.values, 1.0)
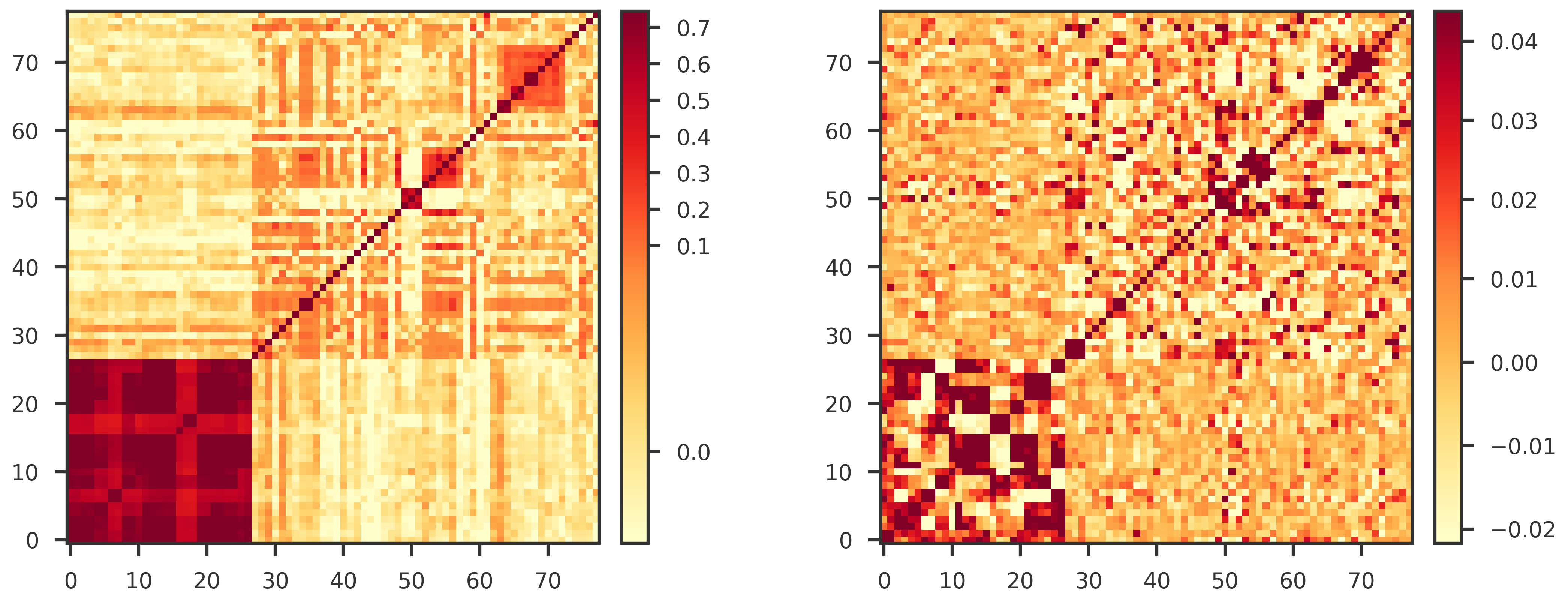
We protected the leading genetic correlation axes of R_g, choosing the smallest number of axes explaining 80% of the positive eigenvalue mass. We remove these axes from the aggressive covariance correction.
Define the genetic-axis projector P = U_k U_k^{\top}, and Q=I-P. Use only the part of noise covariance S in the complement of the genetic axes,
S_{\bot} = QSQ
But S_{\bot} is singular in the full space, so do not directly invert it. Use a shielded precision matrix:
The sampling covariance must be numerically well behaved for the correlated-noise Clorinn objective.
We check for symmetry and positive definiteness.
A symmetric positive-definite covariance is required for stable likelihood evaluation, inversion, and missingness-pattern-specific covariance operations downstream.
Code
tol =1e-8A = shielded_noise_cov.to_numpy()is_symmetric = np.allclose(A, A.T, atol=tol, rtol=0)if is_symmetric:print("This matrix is symmetric.")else:print("This matrix is not symmetric.") max_asymmetry = np.abs(A - A.T).max()print("max_asymmetry:", max_asymmetry)eigvals = np.linalg.eigvalsh((A + A.T) /2)is_pd = np.all(eigvals >0)if is_pd:print ("This matrix is PD.")else:print ("This matrix is not PD.")print (f"Minimum eigenvalue: {eigvals.min():g}")print ("Zero / Negative eigenvalues:")print(eigvals[eigvals <=0])
This matrix is symmetric.
This matrix is PD.
Write release files
The only new release files are shielded_noise_covariance_v1_3.csv and shielded_noise_covariance_v1_4.csv.
Other input files are copied from v1.1 and v1.2 respectively.