This notebook prepares the versioned PGC input files used by the Clorinn v2 pipeline. v1.1 removes biomarker traits, v1.2 additionally removes neurite density traits.
Code
import osimport jsonfrom pathlib import Pathimport numpy as npimport pandas as pdimport matplotlib.pyplot as pltfrom pymir import mpl_stylesheetfrom pymir import mpl_utilsmpl_stylesheet.banskt_presentation(splinecolor='black', dpi=300, colors='kelly')from matplotlib import colormaps as mpl_cmapsimport matplotlib.colors as mpl_colorsfrom mpl_toolkits.axes_grid1 import make_axes_locatable
These are the objects written to disk for downstream Clorinn analyses.
Code
common = A_df.index.intersection(A_df.index).intersection(Z_df.index)# reorder consistentlyA_aligned = A_df.loc[common, common]Z_aligned = Z_df.loc[common, :]# convert to numpyA = A_aligned.to_numpy()Z = Z_aligned.to_numpy()print (f"Number of common traits: {len(common)}")print (f"Number of SNPs: {Z.shape[1]}")
Number of common traits: 51
Number of SNPs: 38281
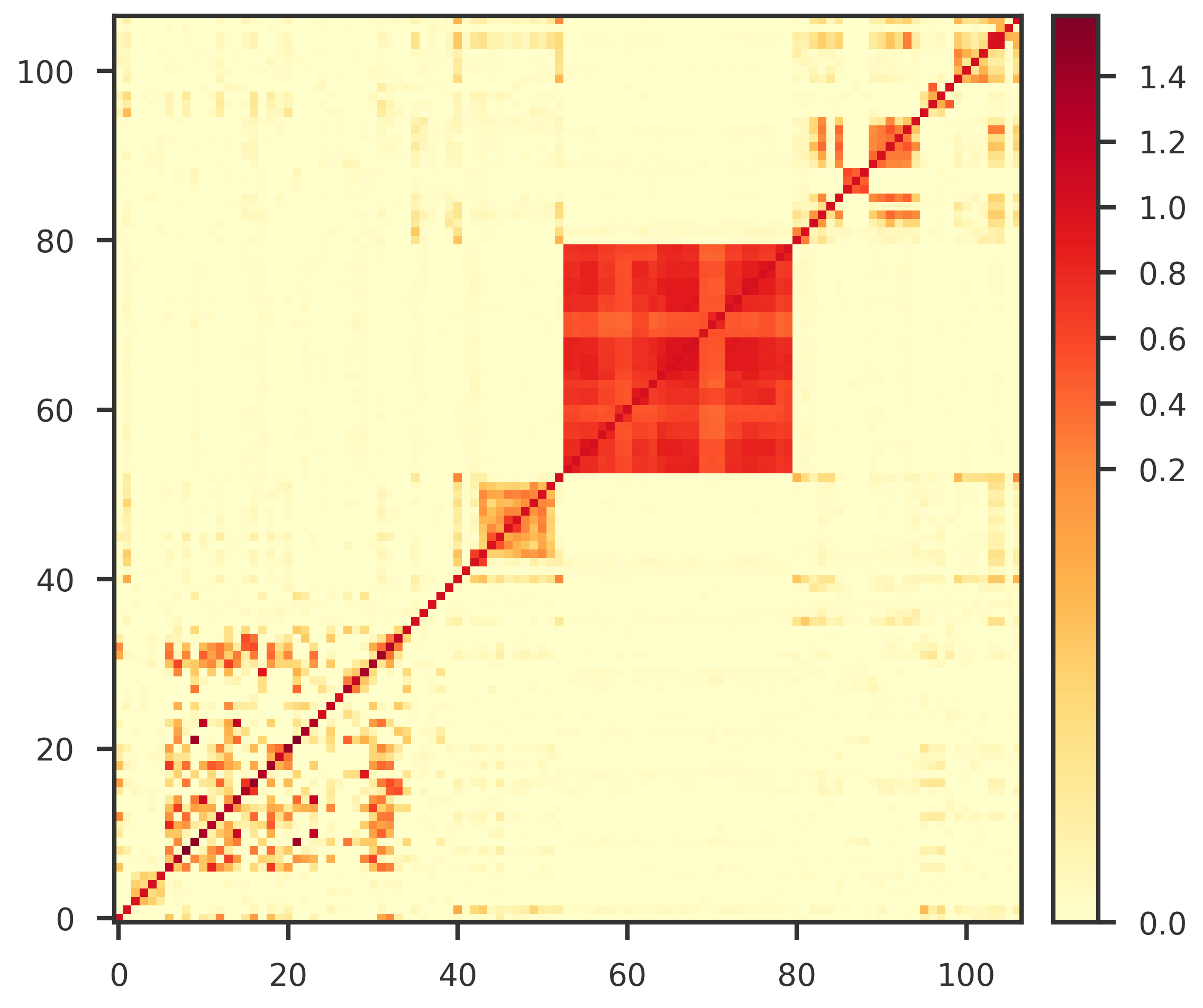
Validate sampling covariance
The LDSC sampling covariance must be numerically well behaved for the correlated-noise Clorinn objective.
We check for symmetry and positive definiteness.
A symmetric positive-definite covariance is required for stable likelihood evaluation, inversion, and missingness-pattern-specific covariance operations downstream.
Code
tol =1e-8is_symmetric = np.allclose(A, A.T, atol=tol, rtol=0)if is_symmetric:print("This matrix is symmetric.")else:print("This matrix is not symmetric.") max_asymmetry = np.abs(A - A.T).max()print("max_asymmetry:", max_asymmetry)eigvals = np.linalg.eigvalsh((A + A.T) /2)is_pd = np.all(eigvals >0)if is_pd:print ("This matrix is PD.")else:print ("This matrix is not PD.")print (f"Minimum eigenvalue: {eigvals.min():g}")print ("Zero / Negative eigenvalues:")print(eigvals[eigvals <=0])
This matrix is symmetric.
This matrix is PD.
Write release files
The final release files are:
Object
Output file
aligned Z-score matrix
zscore_v1_1.csv / ... v1_2.csv
aligned sampling covariance
sampling_covariance_v1_1.csv / ... v1_2.csv
normalized trait-category map
trait_to_group_v1_1.json / ... v1_2.csv
These files define the PGC v1.1 / v1.2 input bundle used by the downstream Snakemake/Clorinn pipeline.
Code
Z_aligned.to_csv(zscore_outfile)A_aligned.to_csv(noise_cov_outfile)withopen(category_outfile, "w") as f: json.dump(trait_to_group, f, indent=4, sort_keys=True)